Small Molecule Drug Design
High-Precision Modeling and Simulations, Real-World Impact
Accelerate discovery cycles with scalable in-silico pipelines that transform complex biology into actionable, high-value candidates.
Identify therapeutic goals and potential binding pockets on target
We begin with a short scoping consult to align on disease biology, clinical context, and decision criteria. Structural assets are curated (experimental or homology models), binding pockets prioritized by druggability and assay readiness, and success metrics defined up front.
Rapid in-silico hit discovery via docking
We model binding sites (and alternate/induced pockets) and run ensemble docking across relevant receptor conformations. Large libraries are screened to yield ranked poses and chemotypes with pose rationales and metadata preserved for downstream refinement.
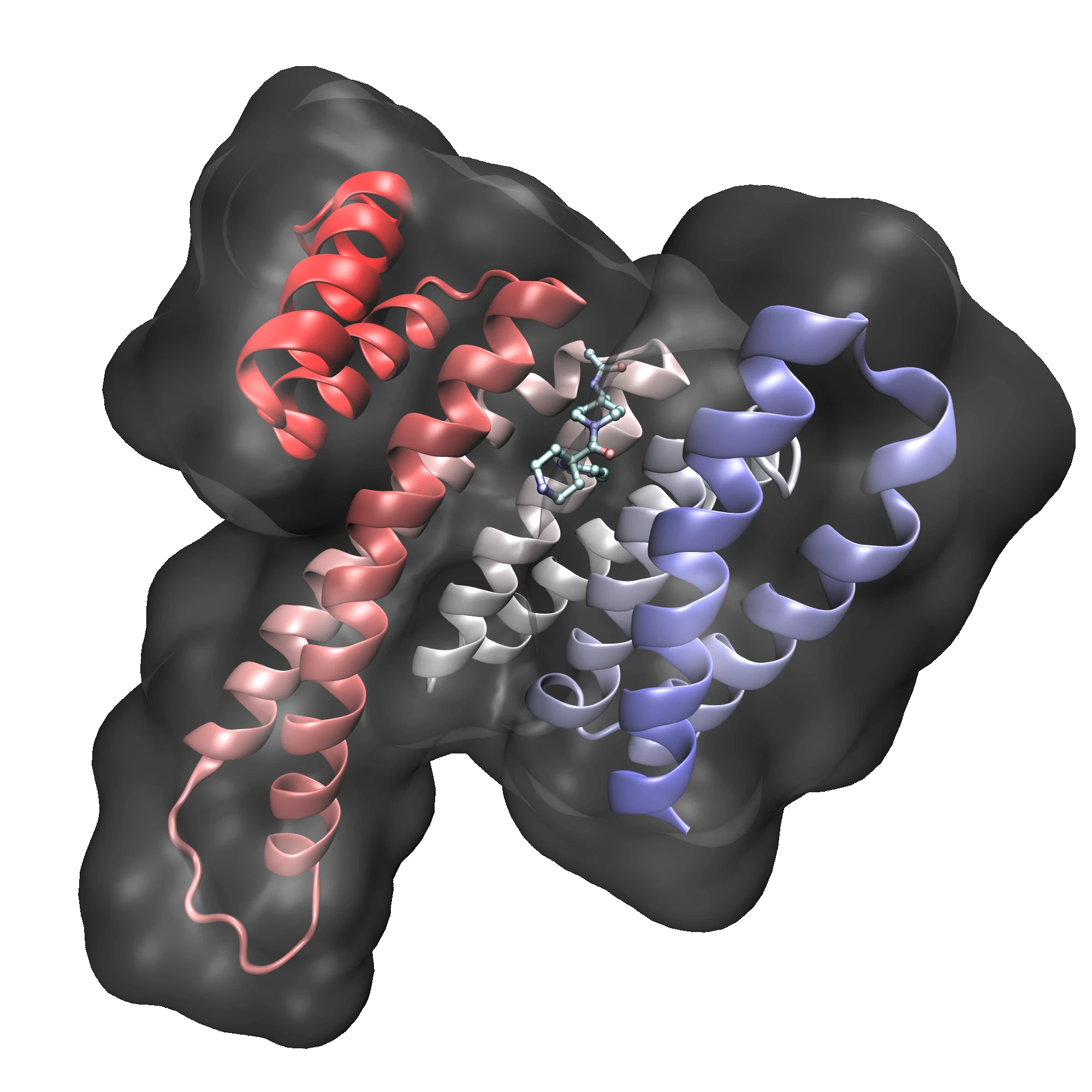
Investigate ligand–target complexes in motion
All-atom MD in explicit solvent relaxes docked poses, captures receptor flexibility, and resolves key water networks. Trajectory clustering and stability metrics (e.g., RMSD, contact/H-bond occupancy) confirm persistent binding modes and filter out unstable complexes.
Quantify ligand-target binding strength
Rigorous free energy methods, such as alchemical free energy perturbation (FEP), thermodynamic integration (TI), or molecular mechanics Poisson-Boltzmann surface area (MMPBSA), are used to quantify binding affinities and provide statistical confidence. This enables rational prioritization of drug candidates before expensive experimental testing.
Guiding chemical modifications to enhance efficacy
Insights from computational simulations are leveraged to elucidate structure-activity relationships and guide chemical modifications for lead compound optimization. A deeper understanding of mechanisms, such as nonspecific binding, can inform future inhibitor design.
Compare pricing
Tailored Solutions. Flexible for Any Budget.
Depending on system size, compute usage & level of support.
Project Scope | 1–2 systems, low–medium complexity | Multiple systems, high complexity | Long-term support, flexible tasks |
Methods | Basic/diverse docking; short–mid MD; µs-level MD; preliminary ML | Extended MD; FEP/TI; DFT; custom ML/workflows | Priority resources; advisory analysis; ad-hoc studies |
Deliverables | Full report + reproducible workflow (includes quick results + summary) | Complete technical dossier + reusable pipeline | Continuous deliverables with monthly milestones |
Use Cases | Feasibility, lead triage, publication-ready prep | Lead optimization, regulatory/Review-ready submissions | Ongoing R&D and parallel projects |
Customizable Report | Fixed Templates |
FAQ
Expert Insights. Scaled to Your Needs
How does the consulting process work?
Our process follows six steps: Scope Determination → Solution Proposal → Pilot Study → Result Presentation → Evaluation → Finalization & Execution. This ensures transparency and alignment at each stage.
What engagement models do you offer?
We offer fixed-price, milestone-based, time-and-materials, and retainer models, depending on project needs and level of support required.
How long does a typical project take?
Timelines depend on complexity, but small pilot studies can be completed in 2–4 weeks, while full-scale projects usually take 2–3 months or more.
Who owns the results and intellectual property?
Clients retain full ownership of results and foreground intellectual property. We work under NDA and provide clear IP agreements.
What types of systems do you work on?
We work across nucleic acids (natural and chemically modified), proteins, small molecules, polymers, and aqueous or complex chemical systems. Our workflows are adaptable to diverse research questions in biology, chemistry, and materials science.
Can you integrate experimental data into the modeling workflow?
Absolutely. Experimental observations such as binding assays, thermodynamic measurements, or structural data can be used to calibrate, benchmark, and validate our computational results.
How reliable are the predictions?
Our results are supported by validation against reference data, convergence diagnostics, and explicit reporting of uncertainties. We emphasize reproducibility and clearly state limitations alongside predictions.
Can you customize workflows for specific problems?
Yes. Every project is tailored to the client’s system, objectives, and available data. We design flexible workflows that balance accuracy, scalability, and cost.
What deliverables will I receive at the end of a project?
Deliverables typically include a detailed report with figures and tables, curated datasets, and reproducible workflows or scripts. All results are prepared to be publication- or presentation-ready.
Do you work with both academic and industry groups?
Yes. We collaborate with academic labs, biotech startups, and established companies worldwide.
Can you support grant or funding applications?
Yes. We provide preliminary computational results, methods descriptions, and figures that can strengthen the technical case of grant or funding proposals.
Can you scale computations using cloud resources if needed?
Yes. We routinely deploy workflows on cloud platforms for large-scale simulations, ensuring cost-efficiency, scalability, and secure data management.
Connect. Collaborate. Grow.
Be part of a growing community of molecular modeling and simulation. Share insights, discuss strategies, and stay updated with the latest trends.